

Vitamins and Nutrients as Primary Treatments
in Experimental Brain Injury: Clinical
Implications for Nutraceutical TherapiesThis section is compiled by Frank M. Painter, D.C.
Send all comments or additions to: Frankp@chiro.org


FROM: Brain Res 2016 (Jun 1); 1640 (Pt A): 114–129 ~ FULL TEXT
Cole Vonder Haar, Todd C. Peterson, Kris M. Martens, Michael R. Hoane
Restorative Neuroscience Laboratory
Department of Psychology,
Life Science II, MC 6502
Southern Illinois University,
Carbondale, IL 62901, USAWith the numerous failures of pharmaceuticals to treat traumatic brain injury in humans, more researchers have become interested in combination therapies. This is largely due to the multimodal nature of damage from injury, which causes excitotoxicity, oxidative stress, edema, neuroinflammation and cell death. Polydrug treatments have the potential to target multiple aspects of the secondary injury cascade, while many previous therapies focused on one particular aspect. Of specific note are vitamins, minerals and nutrients that can be utilized to supplement other therapies. Many of these have low toxicity, are already FDA approved and have minimal interactions with other drugs, making them attractive targets for therapeutics. Over the past 20 years, interest in supplementation and supraphysiologic dosing of nutrients for brain injury has increased and indeed many vitamins and nutrients now have a considerable body of literature backing their use. Here, we review several of the prominent therapies in the category of nutraceutical treatment for brain injury in experimental models, including vitamins (B2, B3, B6, B9, C, D, E), herbs and traditional medicines (ginseng, gingko biloba), flavonoids, and other nutrients (magnesium, zinc, carnitine, omega-3 fatty acids). While there is still much work to be done, several of these have strong potential for clinical therapies, particularly with regard to polydrug regimens.
From the FULL TEXT Article:
Introduction
Traumatic brain injury (TBI) affects 2.5 million individuals in the United States every single year and an estimated 1–2% of the population currently lives with chronic impairments due to TBI [Thurman et al., 1999; Zaloshnja et al., 2008]. In addition to the personal costs associated with brain injury, there is a considerable financial burden associated with primary care, rehabilitation and loss of productivity due to ongoing problems [Humphreys et al., 2013]. Despite the scope of the problem, over 30 years of animal research into the mechanisms and consequences of TBI have failed to yield any successful pharmaceutical agents to treat brain injury in humans. The many unsuccessful clinical trials have caused the field to reconsider several factors involved in clinical and preclinical experimental design. One particular problem with drugs that failed clinical trials is that they were too specific in their treatment targets. This has resulted in a large push in recent years to assess combination therapies, targeting multiple mechanisms of action [Margulies et al., 2015]. As nutritionally-based therapies supplement basic biological function and have therapeutic action in the injured brain, these therapies may eventually represent an important component of combination therapies.
In the clinic, major changes in nutritional status have been observed after TBI. The combination of alterations in blood flow, excitotoxicity, free radical damage and altered global and regional metabolic rates has been identified as a major contributor to secondary damage from brain injury [Vespa et al., 2005]. This metabolic crisis in the early stages of TBI can be detrimental to outcomes and recent studies have shown that supplementing basic nutrition can significantly improve functional outcomes in patients [Horn et al., 2015; Taha et al., 2011]. The guidelines for hospital management of TBI, provided by the Brain Trauma Foundation only include minimal standards for nutritional supplementation, suggesting that patients be placed on full nutritional replacement within 72 h [Bratton et al., 2006]. Of note is that standard nutritional replacement is typically formulated to contain carbohydrates, fats and proteins, with no vitamins or other minerals. Deficiencies in nutrition may further exacerbate TBI symptoms and the depletion of bioactive vitamins, minerals and other compounds may make it difficult for the body to process other pharmaceutical compounds, a phenomenon observed in experimental brain injury [Anderson et al., 2015; Kalsotra et al., 2003].
In this paper, we provide an overview of the overlooked area of nutritionally-based therapies in TBI, focusing on findings at the preclinical level. These therapies, collectively referred to as nutraceuticals, have historically been highlighted as preventative measures for chronic diseases [Lassi et al., 2014; Moyer, 2014; Schleicher et al., 2013]. However, in recent years, many vitamins, minerals and essential nutrients have risen to prominence as potential primary therapeutics and have generated increasing interest [Curtis and Epstein, 2014; Scrimgeour and Condlin, 2014]. Nutraceutical therapies may provide an excellent avenue of treatment for many patients with brain injury. However, they are considerably understudied relative to other pharmacotherapies. The nutrients discussed below represent a wide array of therapeutic mechanisms which offer many opportunities for complementary or even synergistic mechanisms with other pharmaceuticals. Below we highlight the most promising findings from the experimental brain injury literature.
Vitamins
Vitamins are nutrients that are required for normal physiological functioning. Many play crucial roles within the brain in a variety of processes. While vitamins have been widely investigated for their roles in physiology, recent research has begun to examine the how they are involved in dysfunction of the nervous system, from chronic disease to acute insults. The vitamins reviewed below were selected based on existing evidence showing benefits in the treatment of neural insults. A majority of the vitamins have been explored with regards to experimental brain injury, with the exception of vitamins A, B1, B5, B7, B12 and K. Of those that have not been directly assessed in experimental TBI models, vitamin B1 (thiamine) and vitamin B12 (cobalamin) may warrant investigation given that both are important for maintaining nerve function and deficiencies in either have been found to contribute to a variety of neuropathies [Chopra and Tiwari, 2012; Scalabrino and Peracchi, 2006].
Vitamin B2 (Riboflavin)
Riboflavin is a powerful antioxidant acquired from meat and dairy dietary sources. It is readily absorbed, required for normal cellular functioning [Powers, 2003], and has strong antioxidant effects [Hultquist et al., 1993]. Riboflavin rapidly reduces oxidized iron [Hultquist et al., 1993], high levels of which lead to free radical damage and lipid peroxidation [Halliwell and Gutteridge, 1984; Inci et al., 1998]. It delays in vitro neuronal death under excitotoxic conditions in a dose- and time-dependent manner [Lin et al., 2004]. Vitamin B2 is absorbed and phosphorylated to become flavin mononucleotide and is then converted into flavin adenine dinucleotide [Powers, 2003], both of which act as electron carriers in biochemical oxidations and reductions. Additionally, ribo- flavin can be converted to dihydroriboflavin which reduces hemeproteins with high oxidative states of iron, further reducing oxidative damage [Betz et al., 1994; Halliwell and Gutteridge, 1984; Hultquist et al., 1993].
Despite its status as a powerful antioxidant, there have been very few studies of neuroprotection with riboflavin. In experimental brain injury, a dose of 7.5 mg/kg led to substantial functional recovery in sensorimotor function as well as reference and working spatial memory [Barbre and Hoane, 2006; Hoane et al., 2005]. Additionally, animals treated with vitamin B2 had smaller lesions, significant reductions of reactive astrocytes, and less edema. Moreover, in one study, vitamin B2 in combination with magnesium led to fewer impairments and accelerated functional recovery compared to either nutrient alone [Barbre and Hoane, 2006]. While the animal literature is limited, recent clinical studies using a nutrient combination drug that includes riboflavin and nicotinamide (trade name: Cytoflavin) marketed in Russia have shown promise following severe TBI. However, improvement was measured peripherally (e.g., reducing organ failure, sepsis, etc.) and it has not been used to assess neural impairments [Lebedeva et al., 2014]. Vitamin B2 is a strong antioxidant with considerable clinical promise, but further research is needed to validate these findings, at multiple time points, varied dosing parameters, and in additional injury models.
Vitamin B3 (Nicotinamide)
Nicotinamide (NAM) is the amide form of nicotinic acid (niacin) and is currently used clinically in the treatment of pellagra [Yang et al., 2002]. Its mechanism as a neuroprotective agent has been extensively characterized following TBI and stroke [for review, see [Vonder Haar et al. 2013]. The protective actions of NAM are multimodal and include energy supplementation, poly(ADP-ribose)polymerase-1 (PARP) inhibition, free radical scavenging and sirtuin inhibition [Maiese et al., 2009]. NAM increases available energy in the injured brain as a precursor to nicotinamide-adenine dinucleotide (NADţ), which is a critical component of the electron transport chain, assisting in the production of ATP [Maiese and Chong, 2003; Ying, 2008]. The sirtuins and PARP are metabolically-demanding processes which balance the repair of DNA damage and the inhibition of which has been shown to improve outcomes for TBI [Stoica et al., 2014]. Finally, NADţ is a source of free radical scavenging as an electron donor [Maiese et al., 2009]. The combination of these mechanisms has made NAM an attractive target for brain injury therapy.
Vitamin B3 treatment has been shown to be effective across multiple injury models, locations, and doses [Hoane et al., 2006a, 2006b; Hoane et al., 2008b, 2008c]. Specifically, NAM treatment has improved sensory, motor and cognitive function following frontal injury [Hoane et al., 2003, 2008b; Vonder Haar et al., 2011, 2014] and unilateral, sensorimotor cortex injury [Goffus et al., 2010; Hoane et al., 2008c, 2006; Esen et al., 2003; Quigley et al., 2009], with a time window of up to four hours [Hoane et al., 2008b, 2008c]. Further, combination therapy with NAM and progesterone has shown additive effects, including reduced cell death, astrocyte activation, and substantially improved performance in multiple functional assessments [Peterson et al., 2015]. While NAM has shown impressive preclinical efficacy, one study in aged rats observed no benefits, and a trend towards impairment at higher doses [Swan et al., 2011]. Histopathological outcome measures have also demonstrated neuroprotective actions of NAM administration. Acutely (o7 days post injury), vitamin B3 treatment reduced apoptosis, degenerating neurons, edema, and blood-brain barrier compromise, altered the number of activated astrocytes and decreased lesion size [Hoane et al., 2006a, 2006b; Holland et al., 2008]. Chronically (420 days post injury), NAM treatment reduced lesion size and active astrocytes [Goffus et al., 2010; Hoane et al., 2003, 2008b, 2008c, 2006; Peterson et al., 2012; Vonder Haar et al., 2011, 2014]. These neuroprotective effects of NAM are corroborated by brain injury studies specifically examining the downstream targets of NAM, namely the sirtuin receptor, supplementation of NADţ and inhibition of NAD phosphate oxidase, an enzyme involved in oxidative stress [Ansari et al., 2014; Ferreira et al., 2013; Won et al., 2012; Zhao et al., 2012].
The preclinical evidence in rats suggests that NAM may be an interesting treatment to explore in a clinical population. However, there are several problems to consider. A primary concern is that it may have poor actions in aged individuals. With demonstrated, fundamental changes to the NADţ complex during cellular aging [Parihar et al., 2008; Xu and Sauve, 2010], higher levels of NAM may cause toxicity. The reason is not immediately clear, but it is possible that these issues may be associated with actions at sirtuin receptors [Sauve, 2009; Ying, 2008] or changes in free radical scavenging [Li et al., 2006; Ying, 2008]. In addition to the challenge of aged populations, rodent models of TBI have shown that a 50 mg/ kg dose was the minimum to show behavioral effects [Hoane et al., 2008b, 2008c] and that to exhibit maximal recovery, a dose closer to 150–250 mg/kg per day may be necessary [Peterson et al., 2012, 2015; Vonder Haar et al., 2011, 2014]. If this dose was translated directly to humans from the rodent model, it could possibly induce toxic reactions in humans, although doses as high as 80 mg/kg have been tolerated reasonably well [Bussink et al., 2002; Hoskin et al., 1995]. Even considering toxicity issues, NAM may exert protective effects following human TBI and be a particularly interesting target to be used in combination therapies as it is relatively easily administered and has few negative interactions with other drugs.
Vitamin B6 (Pyridoxine)
Vitamin B6 is a water-soluble, readily metabolized and excreted vitamin with relatively low levels of toxicity [Bender, 1999]. It has several different vitamer forms: pyridoxine, pyridoxal, and pyridoxamine, all of which are converted to pyridoxal 50 -phosphate (PLP), primarily in the liver [Hwang et al., 2007; Kelly et al., 2003]. PLP is the active coenzyme of vitamin B6, and is essential for the metabolism, catabolism, and transamination of amino acids [Hwang et al., 2007] as well as several other physiological reactions [Bender, 1999]. It has been suggested that PLP increases the availability of molecules needed for normal metabolic functioning, aids in glycogenolysis [Cabrini et al., 1998; Oka, 2001], and reduces excitotoxicity [Bender, 1999; Roberts et al., 1964], all of which are proposed mechanisms for neuroprotective effects.
There is evidence from the experimental stroke field that PLP is neuroprotective following ischemic injury [Hwang et al., 2007] and that the brain uprgegulates processes involved in PLP production to combat depletion [Hwang et al., 2004]. In experimental brain injury, one study surveyed the effects of a low (300 mg/kg) and intermediate (600 mg/kg) dose of pyridoxine, administered 30 min after unilateral TBI [Kuypers and Hoane, 2010]. Both doses demonstrated some improvements to sensorimotor function, but the higher dose provided increased performance across multiple behaviors. Additionally, only the 600 mg/kg dose demonstrated tissue sparing, suggesting that quite high doses may be necessary to see full benefits. However, chronic high doses of vitamin B6 can cause considerable neural toxicity and behavioral impairments, including balance and gait problems [Krinke et al., 1980; Xu et al., 1989], which limits the feasibility of long-term, high-dose treatment. More work is needed to determine whether an acute dosing paradigm, such as the one described above, would be effective for treating human TBI.
Vitamin B9 (Folic acid)
Folic acid is best known for the role it plays in the closure of the neural tube, but it is also crucial for cell division, DNA synthesis and the maintenance of DNA methylation patterns [Fenech, 2001]. While it has been researched heavily with regards to possible effects on cognition, particularly in the elderly, whether it improves cognitive function is debatable [Fioravanti et al., 1997; Sommer et al., 2003]. Folic acid, along with cobalamin and pyridoxine, is an important cofactor in the homocysteine cycle, which is crucial for a number of processes, including DNA expression and the synthesis of creatine, melatonin and norepinepherine [Miller, 2003]. Any beneficial effects seen in TBI would likely stem from folic acid's action here, since high levels of homocysteine have been shown to induce apoptosis, DNA damage and PARP processes [Kruman et al., 2000]. In experimental brain injury, mild beneficial effects have been observed in the very acute post-injury stage in a piglet model [Naim et al., 2011]. However, these effects were not replicated in a rodent model of TBI, and an increased dose also did not produce any benefit [Vonder Haar et al., 2012]. The few available studies make it difficult to draw conclusions, but generally the benefits of folic acid appear to be minimal in brain injury.
Vitamin C (Ascorbic acid; Ascorbate)
Ascorbic acid is widely recognized as one of the most important endogenous free radical scavengers [Grünewald, 1993]. It has also been suggested to have a neuroprotective role in reducing damage from excitotoxicity [Rice, 2000]. As part of the general metabolic dysfunction in TBI, tissue levels of ascorbic acid have been shown to be severely reduced immediately [Awasthi et al., 1997] and do not return to normal until 72 h post-injury [Tyurin et al., 2000]. Additionally, reduced vitamin C levels have been reported in aged animals as a potential mechanism for increased injury [Moor et al., 2006]. Despite this obvious dysfunction, relatively few studies have attempted direct supplementation of vitamin C. One such study showed that pretreatment with a combination of vitamin C (45–60 mg/kg) and vitamin E preserved ascorbic acid to near sham levels in injured rats and stimulated superoxide dismutase production [Ishaq et al., 2013]. Another study demonstrated preserved motor function and reduced vascular response as a result of vitamin C alone [Wang et al., 2014b]. It is not immediately clear why so few studies have tested ascorbic acid for brain injury. Further study is warranted; however, researchers must be cautious given the limited literature available on the effects of ascorbate in the injured brain.
Vitamin D
Vitamin D is known for its dermal synthesis from cholesterol during sun exposure [Goldblatt and Soames, 1923]. Following synthesis, some is converted the active form, calcitriol, which is carried through plasma to multiple organs via vitamin Dbinding protein [Bouillon et al., 1995]. A large portion of vitamin D's neuroprotective effects are inferred from data on vitamin D deficiency [Hollick, 2007] which suggest that it modulates apoptosis [Thota et al., 2013] and reduces oxidative stress, inflammation and excitotoxicity [Scrimgeour and Condlin, 2014; Tang et al., 2013; Thota et al., 2013]. Deficiencies in vitamin D can contribute to declines in cognitive function, dementia and Alzheimer's disease [Littlejohns et al., 2014]. Additionally, it may act as an antiinflammatory cytokine, dampening immune responses [Adams et al., 2010]. In the experimental brain injury literature, vitamin D is known for its beneficial effects when combined with progesterone [Atif et al., 2013; Hua et al., 2012; Tang et al., 2013] and has recently been extended to a clinical trial [Aminmansour et al., 2015].
In experimental brain injury, vitamin D was initially explored in conjunction with progesterone for its potential to act synergistically, and also to investigate the relationship between age-related decline in vitamin D and brain injury [Cekic and Stein, 2010]. Subsequent studies observed improvements in Morris water maze (MWM) acquisition [Hua et al., 2012] and reduced inflammation and neuronal loss [Tang et al., 2015]. Although effective in adult rats, it appears that this combination may be most beneficial in middle-aged animals, potentially because of existing vitamin D deficiencies. In middle-aged animals, this combination significantly reduced the proliferation of astrocytes, prevented MAP-2 degradation, and reduced neuronal loss [Tang et al., 2013]. The reason for the synergy of vitamin D and progesterone has yet to be fully elucidated, but one study has suggested that it is a combination of reductions in astrocyte activation and NFκB phosphorylation [Tang et al., 2015]. Although more studies are needed to validate vitamin D's additive effects with progesterone under other conditions, there is mounting evidence that the combination of progesterone and vitamins may be a viable follow-up to the failures of progesterone in clinical trials [Aminmansour et al., 2015; Atif et al., 2013; Hua et al., 2012; Peterson et al., 2015; Tang et al., 2013, 2015]. The growing evidence supporting vitamin D, as well as its low toxicity, suggests this vitamin could fill that role. However, further exploration of effects in younger animals, a better understanding of the therapeutic window, and stronger characterizations of functional recovery need to be established prior to moving forward.
Vitamin E
Tocopherols and tocotrienols make up a group of compounds more commonly known as vitamin E, the primary fat-soluble, chain breaking antioxidant in the body [Brigelius-Flohé and Traber, 1999; Parks and Traber, 2000]. The most biologically active form of vitamin E is α-tocopherol (α-T); it is the second most common form of vitamin E in western diets and a lipidsoluble antioxidant which reduces reactive oxygen species [Herrera and Barbas, 2001]. Treatment with vitamin E is effective for some forms of cancer, and prevents and repairs cell tissue damage following radiation [Singh and Krishnan, 2015]. In the central nervous system it has been investigated under lesion [Stein et al., 1991] and TBI models [Clifton et al., 1989; Koc et al., 1999]. The neuroprotective effects of α-T are primarily mediated by its prevention of free radical propagation via the halting of polyunsaturated fatty acid oxidation chain reactions [Burton, 1990; Servet et al., 1998]. It may also have beneficial downstream effects including: altering protein kinase C signaling [Schneider, 2005], decreases in macrophage activation via CD36 signaling [Devaraj et al., 2001], increases of brain derived growth factor [Wu et al., 2010], and decreases in Nogo-A [Yang et al., 2013a].
In models of TBI, α-T combined with polyethylene glycol reduced mortality by 50% and improved motor recovery of function [Clifton et al., 1989]. Similar beneficial effects in cognitive function have been observed with α-T treatment alone, even when administered up to 90 days after injury [Stein et al., 1991; Wu et al., 2010; Yang et al., 2013b]. Furthermore, vitamin E reduces amyloidosis and improves cognitive function after repetitive TBI in a model of Alzheimer's disease [Conte et al., 2004]. Although these behavioral effects are substantial, one study has shown limited efficacy of vitamin E on lipid peroxidation in the acute post-injury phase [Koc et al., 1999], but others have highlighted improvements in markers of oxidative stress at later time points [Ishaq et al., 2013; Servet et al., 1998]. Additional studies have demonstrated that extended pretreatment confers the strongest reductions in lipid peroxidation and oxidative stress [Hall et al., 1992; Wu et al., 2010]. The preclinical data supporting vitamin E in TBI are strong, especially considering that there is a significant drop in plasma and brain levels of vitamin E following injury [Ishaq et al., 2013]. Further, the pharmacology in humans is known and is considered relatively safe in its use as an anticonvulsant [Spiegel and Noseworthy, 1963]. While it has high lipid solubility and low toxicity [Clifton et al., 1989; Veinbergs et al., 2000], it takes a considerable amount of time to reach effective levels in the CNS [Hall et al., 1992] and can cause hemorrhage at very high doses [Sesso et al., 2008]. These limitations should be taken into account when considering vitamin E either alone or in a polytherapy for patients.
Herbs and traditional Chinese medicines
Herbal remedies have been used in many cultures for a variety of medicinal purposes, ranging from dubious benefits to effective treatments for crippling disorders. Though a large portion of this medicine is comes from tradition rather than evidence-based approaches, there is a considerable amount of research emerging on specific medical benefits of the chemicals found in many herbs and roots. Of interest for those studying brain injury are the herbs that may affect aspects of the secondary cascade, namely those with antioxidant, anti-apoptotic or neuroprotective effects.
Ginseng
Ginseng is a family of herbs that has been used in traditional Chinese medicine for many centuries. Though it is a plant with many complex molecules, several bioactive components have been identified. The primary class is a chemical group called saponins, of which the ginsenosides are the most important. Recently, ginseng has gained attention as a preventative for varied conditions such as influenza, cancer and even impaired cognition [Lee et al., 2008; Scaglione et al., 1995; Yun et al., 2010]. Interestingly, the common link between many of these diseases is inflammation, which ginseng has been shown to reduce [Lee and Lau, 2011]. In experimental TBI studies, combined saponins from ginseng have been shown to improve a variety of behavioral functions, both cognitive and motor in a dose-dependent fashion, with doses of 200 mg/kg showing the greatest benefits [Hu et al., 2014; Ji et al., 2005; Kumar et al., 2014a; Xia et al., 2012]. One study even found improvements when ginseng was administered 15 days after the initial injury [Kumar et al., 2014a]. These studies also found histopathological improvements: ginseng reduced markers of oxidative stress and inflammation [Kumar et al., 2014a; Xia et al., 2012], decreased cell loss [Hu et al., 2014; Ji et al., 2005; Xia et al., 2012] and reduced apoptosis [Xia et al., 2012]. Studies in TBI have yet to evaluate the specific ginsenosides responsible for these beneficial effects, but there is a robust literature in the field of experimental stroke for researchers interested in this topic. The results from these studies suggest that ginseng may provide neuroprotection through a combination of antiinflammatory and antioxidant mechanisms.
Gingko biloba
G. biloba is a tree that dates back to prehistoric times, the leaf extract of which is commonly available as an over-thecounter supplement. The extract form contains several compounds, including flavonoids (see section below) as well as ginkgolides, which are likely the bioactive components [Diamond et al., 2000]. Gingko has not been widely used in the treatment of brain injury, however it has been explored as a treatment for diseases related to TBI, including Alzheimer’s disease, with some beneficial effects observed [Yang et al., 2015]. One study has specifically assessed treatment of experimental TBI by G. biloba extract and observed improvements in motor and cognitive function. Treatment also reduced cell loss in multiple regions of the brain, but failed to improve the immediate lesion cavity [Hoffman and Stein, 1997]. Another study utilized ginkgolide B, a substrate of the plant, and observed reductions in apoptosis and inflammatory markers [Yu et al., 2012]. Although much more evidence is needed to determine the efficacy of ginkgo in TBI, there are other promising studies regarding ginkgo and ischemic injury [Mdzinarishvili et al., 2012; Yang et al., 2013b; Zhang et al., 2012].
Flavonoids
Flavonoids are plant metabolites with many common dietary sources, including fruits, vegetables, teas and wine. They serve primarily as antioxidant agents, reducing free radicals in tissues [Heim et al., 2002]. Because of this, high dietary intake of flavonoids is associated with reduced risk for a number of diseases, including heart and cerebrovascular disease, diabetes and some types of cancer [Knekt et al., 2002]. The brain injury field has taken note of these mechanisms and recently a number of laboratories have begun assessing the efficacy of the different flavonoids to treat experimental TBI. Further, Enzogenol, a bark extract containing multiple flavonoids, has already been assessed in a phase II clinical trial, in which it was deemed safe and suggested to accelerate recovery from mild TBI [Theadom et al., 2013].
Due to the many types of flavonoids, several different compounds have been assessed in animal models of TBI. However, not all have had repeated assessment across multiple labs, limiting the generalization of the findings for specific flavonoids. Most flavonoids have potent antioxidant properties and work to improve redox status; through this, they indirectly reduce neuroinflammation as well. In experimental TBI, luteolin has received the most attention for its ability to reduce a variety of markers of oxidative stress, inhibit apoptosis, reduce inflammation and decrease edema [Cordaro et al., 2014; Sawmiller et al., 2014; Xu et al., 2014a, 2014b]. Interestingly, one study using transgenic Alzheimer mice, demonstrated that luteolin administration prevented TBI-induced upregulation of beta-amyloid, phosphorylated tau and glycogen synthase kinase-3 [Sawmiller et al., 2014]. One study suggested that the effects of luteolin are primarily mediated through the Nrf2 pathway [Xu et al., 2014a] and another suggested that increased autophagy may account for other protective effects [Xu et al., 2014b]. Unfortunately, only minimal motor testing has been performed to assess functional recovery using luteolin [Cordaro et al., 2014; Xu et al., 2014a], and more will be needed to determine the efficacy of this drug. Quercetin is another antioxidant flavonoid that has been shown to improve cognitive performance in the MWM and normalize firing rates of neurons in injured brains [Schültke et al., 2005; Yang et al., 2014]. Further, markers of oxidative stress, inflammation and apoptosis were also reduced [Yang et al., 2014]. Pycnogenol, a commercially available supplement, reduced oxidative stress, inflammatory cytokines and improve markers of synaptic function after injury [Ansari et al., 2013; Scheff et al., 2013]. Several other antioxidant flavonoids for TBI have also been evaluated, albeit only in single studies. Baicalein, puerarin and formononentin improved oxidative status and reduced cell death [Li et al., 2014b; Wang et al., 2014a], as well as reduced inflammatory markers and improved sensorimotor function [Chen et al., 2008; Li et al., 2014b].
Other flavonoids appear to have distinct actions apart from antioxidant properties. Of specific note is 7,8-dihydro- flavone (7,8-DHF), which stimulates growth factors through activation of the TrkB BDNF receptor. Treatment with 7,8-DHF has been shown to improve markers associated with learning and plasticity, specifically by preventing TBI-induced cell death of new neurons and by rescuing phosphorylated creb and GAP-43 levels [Agrawal et al., 2015; Chen et al., 2015]. These actions improved spatial memory, even when the drug was administered several days after injury [Agrawal et al., 2015]. Another pair of flavonoids have demonstrated direct anti-inflammatory action in TBI models. Wogonin has been shown to reduce inflammation through a TLR4-mediated pathway, leading to improved behavioral function and reduced cell death and cavitation [Chen et al., 2012]. Flavopiridol, as a cell-cycle inhibitor, directly inhibits activation of microglia and astrocytes, causing smaller lesion volume, less glial scarring and providing recovery on motor and cognitive behaviors [Di Giovanni et al., 2005]. The various flavonoids have strong potential for the treatment of TBI, however, given the variety of substances, much more research is needed to identify common pathways by which they exert their effects and determine which are the most effective for TBI.
Other nutrients
Magnesium
Over the last several decades, a large body of evidence has accumulated suggesting that Mg2ţ is vitally important in various neurological injuries and that it interacts with other micronutrients to maintain and promote cognitive function and performance [Huskisson et al., 2007]. In particular, the role that Mg2ţ plays in the pathophysiological processes following traumatic brain injury (TBI) and the efficacy of Mg2ţ therapy in promoting functional recovery across a variety of animal models has been well demonstrated [Hoane, 2004; Hoane and Barth, 2001; Sen and Gulati, 2010; Van Den Heuvel and Vink, 2004; Vink et al., 2009]. Mg2ţ has been shown to be effective in preventing excitotoxic damage involved in a variety of types of neural damage and is also involved in regulating antioxidant capabilities, particularly in the aging brain [Barbagallo and Dominguez, 2010; Vink and McIntosh, 1990]. The importance of Mg2ţ in normal cellular functioning has been well documented, as has its importance in the pathophysiology following injury. Previously, several reviews addressing these issues have been written [Hoane, 2004; Hoane and Barth, 2001; Sen and Gulati, 2010; Van Den Heuvel and Vink, 2004] so the mechanistic actions will not be chronicled here; instead, the focus will be on functional outcome studies.
The use of Mg2+ therapies to promote recovery of function has been investigated for several decades. Treatment with magnesium has been used in models of ischemia [Izumi et al., 1991; Tsuda et al., 1991; Vacanti and Ames, 1984], focal cortical lesions [Hoane et al., 2000, 1998, 1997; Hoane and Barth, 2001, 2002], and spinal cord injuries [Kwon et al., 2010] to highlight but a few of many studies. In experimental TBI, previous work has identified that dietary deficiencies in Mg2ţ lead to poorer functional outcomes and increased cell death; however, some of these deficits can be rescued by Mg2ţ administration and supplementation post injury [Heath and Vink, 1999; Hoane et al., 2008a; McIntosh et al., 1988]. Furthermore, at doses between 150–1000 mm/kg, Mg2ţ administration in animals with normal diets causes improvements in sensorimotor functioning, memory and decreases in anxiety following TBI [Enomoto et al., 2005; Hoane, 2005; McIntosh et al., 1989; Vink et al., 2003]. In addition, these animals demonstrate reductions in a variety of histopathological outcomes including glial proliferation, BBB breach, edema and neuronal death [Enomoto et al., 2005; Esen et al., 2003; Ghabriel et al., 2006; Park and Hyun, 2004]. Collectively, these findings suggest that Mg2ţ modifies recovery of function following neurological injury and that dietary magnesium may reduce the subsequent risks of such injuries. Although there have been recent failed clinical trials for both TBI and stroke [Saver et al., 2015; Temkin et al., 2007], further research is warranted with regards to combination therapies. Future studies should focus on using Mg2ţ to augment the existing effects of other pharmaceuticals and examine strategies for rapidly increasing brain concentrations of Mg2ţ.
Zinc
Zinc holds a controversial role in TBI pathophysiology. Numerous studies have identified increased, toxic levels of zinc following experimental injury, yet others have highlighted zinc deficiency as a major problem after TBI and demonstrated zinc supplementation to be an effective therapy. It has been repeatedly suggested that zinc may contribute to excitotoxic cell death [Frederickson et al., 2005, 2004] and studies in TBI have linked zinc accumulation to cell death [Hellmich et al., 2007; Suh et al., 2000]. A likely candidate for zinc damage in TBI is that cell death due to excitotoxicity releases excess zinc, which is normally highly protein-bound (80%). This free zinc then interferes with cell processes via oxidative mechanisms, mitochondrial interference and MAPK-related cell death pathways [Lau and Tymianski, 2010]. Because of this, removal of excess zinc, via chelation or targeted chemicals has been evaluated across several studies with a mixture of beneficial [Hellmich et al., 2004; Suh et al., 2000], null [Choi et al., 2014; Hellmich et al., 2008] and detrimental results [Doering et al., 2010].
Because patients have shown zinc deficiency following TBI [McClain et al., 1986], zinc supplementation has been evaluated in both patients and rats. Results in patients showed a trend towards improvements [Young et al., 1996] and in rats, zinc provided moderate improvements to function [Cope et al., 2012, 2011]. Further, zinc deficiency in rodents has exacerbated neural injury [Yeiser et al., 2002]. The mechanism by which zinc may exert its neuroprotective actions is not well understood; however, there are several likely candidates. One possibility is that zinc may affect redox signaling directly [Li et al., 2010], however other studies call into question whether this action is beneficial or detrimental [Bishop et al., 2007]. Given the extreme mix of results regarding zinc, researchers will need to carefully evaluate the potential effects, both beneficial and detrimental of using this as a therapy.
Carnitine
Normal mitochondrial function requires the amino acid derivative carnitine, of which, the active stereoisomer is acetyl-L-carnitine (ALC). ALC is synthesized in the brain [Jones et al., 2010] and is also commercially available as a supplement at nutrition retailers. It is easy to administer, crosses the blood-brain barrier [Kido et al., 2001] and has low toxicity [Wainwright et al., 2003]. Following neural insult, mitochondrial respiration and energy production are altered. Multiple studies have examined ALC's ability to repair mitochondrial function and improve functional recovery after hypoxic ischemia [Rosenthal et al., 1992; Slivka et al., 1990; Wainwright et al., 2003] glutamate-induced excitotoxicity [Nagesh Babu et al., 2011], as well as brain [Scafidi et al., 2010] and spinal cord injury [Azbill et al., 1997; Conta Steencken and Stelzner, 2010; Patel et al., 2012, 2010; Sullivan et al., 2003; Yu et al., 2009]. The specific mechanism by which ALC exerts its effects is unknown, but it is likely to involve increases of ATP through the NADHţ mediated electron transport chain and reductions of high levels of acyl-CoA esters that can impair mitochondrial processes [Scafidi et al., 2010]. Additionally, it may mediate cellular stress responses by inducing heat-shock proteins to repair and prevent damage [Calabrese et al., 2006].
Many studies of spinal cord injury have utilized ALC to improve mitochondria function and demonstrated critical neuroprotection [McEwen et al., 2011; Patel et al., 2010, 2009; Springer et al., 2010; Xiong et al., 2009]. Despite these findings in a closely-related field, only one has assessed the effects of carnitine in TBI. The researchers observed improvements to near-sham levels in motor and cognitive functioning early after injury and lesion volumes were significantly reduced [Scafidi et al., 2010]. While the animal studies are limited, a human study (nonrandomized, open-label) in retired National Football League players used ALC as part of a combination therapy which improved performance and brain perfusion in players who received multiple TBIs [Amen et al., 2011]. Additionally, it has been used in the clinical treatment of Alzheimer's disease, depression, age, diabetes, ischemia and other neurological diseases specifically associated with metabolic compromise [Bonavita, 1986; Onofrj et al., 1995; Rai et al., 1990; Spagnoli et al., 1991; Tempesta et al., 1987]. One potential concern is that the majority of experimental studies on ALC were performed in the immature brain [Patel et al., 2010; Scafidi et al., 2010; Wainwright et al., 2003] and more research needs to be completed to determine if ALC is effective in other populations. Additional experiments are required to explore sex differences, and determine whether the therapeutic window can be extended beyond 1-h postinjury [Scafidi et al., 2010]. Although the safety index and beneficial effects of ALC is promising, research supporting its effects following TBI is still in its infancy.
Omega-3 Fatty Acids
Omega-3 acids are polyunsaturated fats found in both plants and fish and have received much attention regarding prevention of cancer, heart disease and stroke, although the scope of these effects are debated [Campbell et al., 2013]. They play a varied role in the CNS, providing a substrate for neuronal membrane phospholipids, modulating neurotransmission, and protecting cells from oxidative stress and inflammation through metabolites [Niemoller et al., 2009]. These acids have been a subject of interest in the field of TBI for several years, particularly with regard to their use as a prophylactic treatment. Multiple recent reviews have emphasized the potential for these in TBI [Hasadsri et al., 2013; Michael-Titus and Priestley, 2014], thus this section will only briefly discuss their putative mechanism and potential. Omega-3 acids in brain injury are thought to act by two primary mechanisms, but potentially have numerous other effects as well. First, they modulate neuronal survival by preventing axonal loss after injury. This occurs by increasing BDNF levels, reducing oxidative stress, and preventing synapse degradation [Kumar et al., 2014b; Wu et al., 2004]. Second, they are strong anti-inflammatory agents, actively reducing pro-inflammatory cytokines such as TNF-α, IL-6, and C-reactive protein and promoting the clearance of neutrophils [Ferrucci et al., 2006; Li et al., 2014a]. In addition to these mechanisms, there are several suggested effects with less evidence. Notably, AMPA receptor modulation may reduce levels of excitotoxicity as well as regulation of ion channels and Ca2ţ pumps which may also reduce excitotoxicity and other problems associated with energy deficiency after brain injury [Ménard et al., 2009; Vreugdenhil et al., 1996].
While the biochemical evidence is quite promising, there are relatively few studies that have examined functional outcomes associated with omega-3 acids and brain injury. Previous studies have examined the effects of modulating fatty acids prior to injury. One such study found that depletion of omega-3 acids led to worsened motor and memory deficits [Desai et al., 2014] and others have shown that supplementation prior to injury leads to improvements in motoric ability and learning [Pu et al., 2013; Wang et al., 2013; Wu et al., 2004]. Despite these promising results, a clinical trial examining fish oil and other compounds after injury found no improvements in mortality from brain injury, but did see improvements in some peripheral issues (e.g. infections) [Painter et al., 2015]. The cumulative evidence regarding omega 3 fatty acids is quite promising in the treatment of brain injury, however further investigation is needed. One of the biggest considerations is whether fish oils are only effective as a prophylactic treatment. While this may limit the applicability of these in the general populace, in vulnerable populations such as athletes or military personnel, omega-3 acids could provide strong benefits given the ease of integrating them into diet.
Discussion
There is a robust, yet disparate literature emerging on treatments using nutritionally-based therapies for the treatment of experimental brain injury. The largest challenges facing these therapies are similar to those in other areas of treatment, namely the need for replication and verification of effects and disinterest from pharmaceutical companies. Several of the nutraceuticals discussed above have evidence stemming primarily from a single laboratory (e.g., Bvitamins – Hoane laboratory, vitamin D – Stein laboratory). This underscores the need for additional research to verify effects in other models of brain injury and under other laboratory conditions to determine how truly translational these therapeutics are. Additionally, it is unclear whether some of these compounds are understudied (e.g. vitamin B6, vitamin B9, vitamin C) or whether, due to publication bias, neutral or negative results have not been published. The lack of clinical interest in many of these treatments is primarily a monetary issue. It is difficult to convince pharmaceutical companies to develop a drug that cannot be patented. There are some ways to work around this problem, and the clinical development of progesterone is good evidence for this [Skolnick et al., 2014]. However, the best solution would be for federal funding to explore treatment options that are difficult to patent.
Despite these concerns, many of which apply to any therapeutics being evaluated for brain injury, there is considerable promise in a number of nutritionally-based therapies given the current preclinical evidence. In particular, nicotinamide, magnesium, the flavonoids, and omega-3 acids have a broad body of research supporting their use in the treatment of TBI. Nicotinamide has potent neuroprotective effects through its multimodal mechanisms of supporting energy production, inhibiting PARP activity and free radical scavenging [Maiese et al., 2009]. This has been borne out through studies of both experimental stroke and TBI over the course of many years [Hoane et al., 2003; Peterson et al., 2015; Vonder Haar et al., 2014; Yang et al., 2002]. Further, the timewindow for recovery of function has been shown to be around 4 h in the rat [Hoane et al., 2008b, 2008c], which may fit into the timeframe for the treatment of human injuries. Magnesium, while primarily acting on only one target, excitotoxicity, has very strong effects in attenuating damage and providing functional recovery [Hoane, 2005; McIntosh et al., 1989; Vink et al., 2003]. Unfortunately, recent failures in clinical trials indicate that is not efficacious on its own and may limit interest. However, in combination with treatments targeting other aspects of the secondary damage cascade, there is still considerable potential for magnesium. The flavonoids are a diverse class of molecules, possessing strong antioxidant, anti-inflammatory and even growth factor-stimulating properties [Agrawal et al., 2015; Chen et al., 2012; Schültke et al., 2005]. There is converging evidence from multiple laboratories and on the benefits of these in treating brain injury. The large drawback to treating brain injury with antioxidant agents is that many require very early administration for full efficacy [RodriguezRodriguez et al., 2014], however one, 7,8-DHF, has shown improvements even when administered days after injury [Agrawal et al., 2015]. Finally, omega-3 acids have shown large potential in the prophylactic treatment of TBI [Hasadsri et al., 2013; Pu et al., 2013] and have broad mechanisms of action that affect several points in the secondary injury cascade, including inflammatory signaling and cellular plasticity [Li et al., 2014a; Wu et al., 2004].

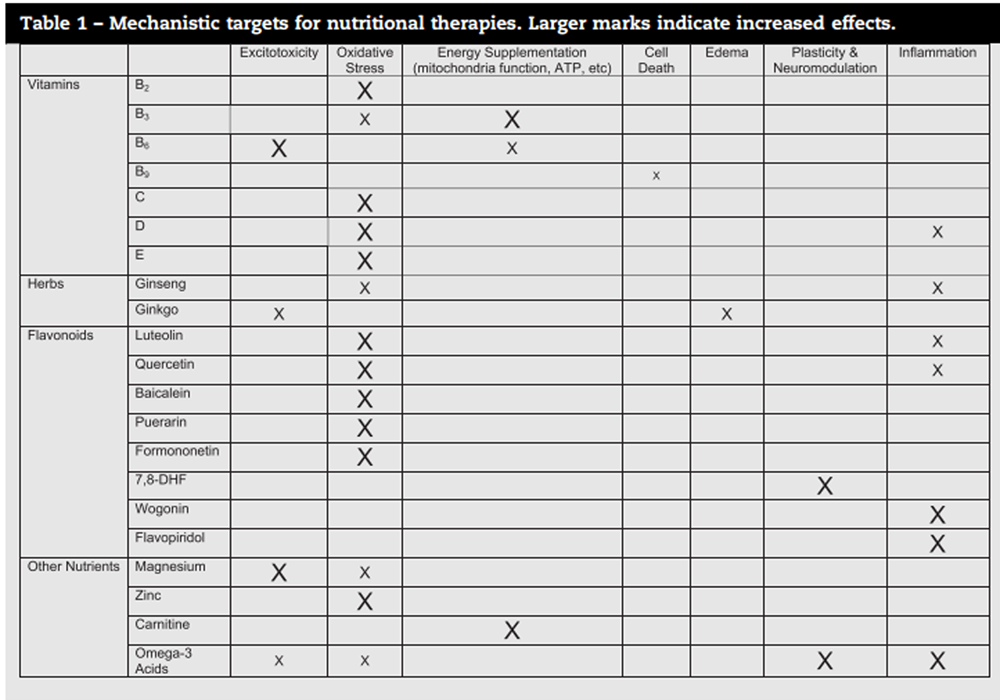
Table 1 It is unlikely that any of these treatments will be successful on their own in treating human brain injury. The biggest potential for all of the therapies discussed in this review is in combination therapy. While several of these have multimodal action on different aspects of the secondary injury cascade, none of them address all of the issues with brain injury (see Table 1 for a summary of mechanisms). Researchers interested in combining therapies such as these are advised to consider treatments with complementary mechanisms of action in order to provide additive or synergistic benefit. The vitamins and nutrients reviewed above have a variety of mechanisms, meaning they could readily be combined with each other or with existing pharmaceuticals in development. In the antioxidant category, numerous flavonoids, ginseng and vitamins B2, C, D, and E all have demonstrated beneficial effects. For excitotoxicity, the options are more limited, but magnesium provides relatively strong effects in blocking excitotoxic damage, and vitamin B6 may also have potential in this area. Two agents, vitamin B3 and carnitine, are effective neuroprotectants through their mechanism of energy supplementation. Additionally, several flavonoids and omega-3 acids improve neuroinflammatory status. Finally, the flavonoid 7,8-DHF and omega-3 acids improve function through other mechanisms such as stimulating growth factors. While toxicity needs to be monitored as nutrients are combined and used in very high doses, many of these have limited toxicity and are likely to have minimal interactions with other agents. This, combined with their diverse mechanisms of action could make them quite beneficial for inclusion in polydrug treatments.
References:
Please refer to the Full-Text article 
Return to NUTRITION
Return to OMEGA-3 FATTY ACIDS
Since 1-04-2015
| Home Page | Visit Our Sponsors | Become a Sponsor |
Please read our DISCLAIMER |